

Video could not be played
Research
Our work is focused on the expression and travels of RNA within the cell, from the site of its birth to its ultimate biological destiny in the cytoplasm where it makes proteins in specific locations. The following excerpts from our recent publications highlights the lab's research interests, which include: gene expression, transcription, transport, localization & translational regulation, degradation, reagent development and microscopy development.
Gene Expression from
Transcription of functionally related constitutive genes is not coordinated.
Gandhi S, Zenklusen D, Lionnet T, Singer RH.
Nat Struct Mol Biol 18(1):27-34 (2011 January). PMCID: PMC3058351
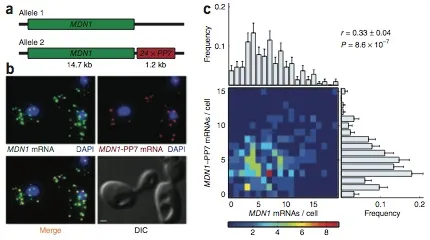
Expression of an individual gene can vary considerably among genetically identical cells because of stochastic fluctuations in transcription. However, proteins comprising essential complexes or pathways have similar abundances and lower variability. It is not known whether coordination in the expression of subunits of essential complexes occurs at the level of transcription, mRNA abundance or protein expression. To directly measure the level of coordination in the expression of genes, we used highly sensitive fluorescence in situ hybridization (FISH) to count individual mRNAs of functionally related and unrelated genes within single Saccharomyces cerevisiae cells. Our results revealed that transcript levels of temporally induced genes are highly correlated in individual cells. In contrast, transcription of constitutive genes encoding essential subunits of complexes is not coordinated because of stochastic fluctuations. The coordination of these functional complexes therefore must occur post- transcriptionally, and likely post-translationally.
Video could not be played
Correlation between transcripts from two alleles of a constitutively active gene, MDN1, in diploid cells.
Localization & Translational Regulation from
Spatial arrangement of an RNA zipcode identifies mRNAs under post-transcriptional control.
Patel VL, Mitra S, Harris R, Buxbaum AR, Lionnet T, Brenowitz M, Girvin M, Levy M, Almo SC, Singer RH, Chao JA.
Genes Dev 26(1):43-53 (2012 January 1). PMCID: PMC3258965
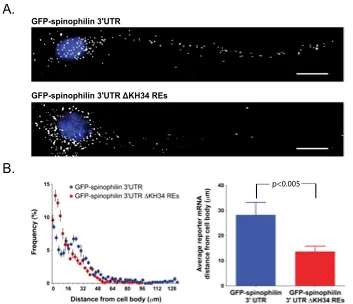
How RNA-binding proteins recognize specific sets of target mRNAs remains poorly understood because current approaches depend primarily on sequence information. In this study, we demonstrate that specific recognition of messenger RNAs (mRNAs) by RNA-binding proteins requires the correct spatial positioning of these sequences. We characterized both the cis-acting sequence elements and the spatial restraints that define the mode of RNA binding of the zipcode-binding protein 1 (ZBP1/IMP1/IGF2BP1) to the β-actin zipcode. The third and fourth KH (hnRNP K homology) domains of ZBP1 specifically recognize a bipartite RNA element comprised of a 5' element (CGGAC) followed by a variable 3' element (C/A-CA-C/U) that must be appropriately spaced. Remarkably, the orientation of these elements is interchangeable within target transcripts bound by ZBP1. The spatial relationship of this consensus binding site identified conserved transcripts that were verified to associate with ZBP1 in vivo. The dendritic localization of one of these transcripts, spinophilin, was found to be dependent on both ZBP1 and the RNA elements recognized by ZBP1 KH34.
Video could not be played
The ZBP1 KH34 REs are required for distal dendritic RNA transport.
Degradation from
Single-Molecule mRNA Decay Measurements Reveal Promoter-Regulated mRNA Stability in Yeast.
Trcek T, Larson DR, Moldón A, Query CC, Singer RH.
Cell 147(7):1484-1497 (2011 December 23).
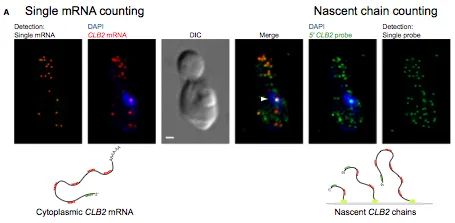
Messenger RNA decay measurements are typically performed on a population of cells. However, this approach cannot reveal sufficient complexity to provide information on mechanisms that may regulate mRNA degradation, possibly on short timescales. To address this deficiency, we measured cell cycle-regulated decay in single yeast cells using single-molecule FISH. We found that two genes responsible for mitotic progression, SWI5 and CLB2, exhibit a mitosis-dependent mRNA stability switch. Their transcripts are stable until mitosis, when a precipitous decay eliminates the mRNA complement, preventing carryover into the next cycle. Remarkably, the specificity and timing of decay is entirely regulated by their promoter, independent of specific cis mRNA sequences. The mitotic exit network protein Dbf2p binds to SWI5 and CLB2 mRNAs cotranscriptionally and regulates their decay. This work reveals the promoter-dependent control of mRNA stability, a regulatory mechanism that could be employed by a variety of mRNAs and organisms.
Video could not be played
Measuring mRNA Decay Rates Using Single-Cell, Single-Molecule FISH.
Regent Development from
A transgenic mouse for in vivo detection of endogenous labeled mRNA.
Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, Wells AL, Chao JA, Park HY, de Turris V, Lopez-Jones M, Singer RH.
Nat Methods 8(2):165-170 (2011 February). PMCID: PMC3076588
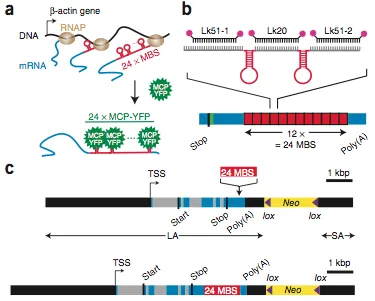
live-cell single mRNA imaging is a powerful tool but has been restricted in higher eukaryotes to artificial cell lines and reporter genes. We describe an approach that enables live-cell imaging of single endogenous labeled mRNA molecules transcribed in primary mammalian cells and tissue. We generated a knock-in mouse line with an ms2 binding site (MBS) cassette targeted to the 3′ untranslated region of the essential ß-actin gene. As ß-actin-MBS was ubiquitously expressed, we could uniquely address endogenous mRNA regulation in any tissue or cell type. We simultaneously followed transcription from the ß-actin alleles in real time and observed transcriptional bursting in response to serum stimulation with precise temporal resolution. We tracked single endogenous labeled mRNA particles being transported in primary hippocampal neurons. the MBS cassette also enabled high-sensitivity fluorescence in situ hybridization (FISH), allowing detection and localization of single ß-actin mRNA molecules in various mouse tissues.
Video could not be played
Schematic of the Actb-MS2 system for live-cell imaging.
Microscopy Development from
Calibrating excitation light fluxes for quantitative light microscopy in cell biology.
Grünwald D, Shenoy SM, Burke S, Singer RH.
Nat Protoc 3(11):1809-1814 (2008 November). PMCID: PMC3109976
Power output of light bulbs changes over time and the total energy delivered will depend on the optical beam path of the microscope, filter sets and objectives used, thus making comparison between experiments performed on different microscopes complicated. Using a thermocoupled power meter, it is possible to measure the exact amount of light applied to a specimen in fluorescence microscopy, regardless of the light source, as the light power measured can be translated into a power density at the sample. This widely used and simple tool forms the basis of a new degree of calibration precision and comparability of results among experiments and setups. Here we describe an easy-to-follow protocol that allows researchers to precisely estimate excitation intensities in the object plane, using commercially available opto-mechanical components. The total duration of this protocol for one objective and six filter cubes is 75 min including start-up time for the lamp.

Video could not be played
Beam profile along the optical axis of the microscope.