BACKGROUND
Autoimmune Thyroid Diseases (AITD)
(Reviewed in Hasham, Tomer. Immunol Res 2012; 54: 201-213)
Autoimmune thyroid diseases (AITD), include Graves’ disease (GD) and Hashimoto’s thyroiditis (HT). AITD are reported to be the commonest autoimmune endocrine disorders, affecting up to 5% of the population (for review see Huber, et al. Endocr Rev 2008; 29: 697-725). Both conditions are characterized pathologically by infiltration of the thyroid by T- and B-cells reactive to thyroid antigens with production of thyroid specific autoantibodies. However, despite the similar pathophysiology of GD and HT the result is two clinically opposing syndromes. In GD, lymphocytic infiltration leads to activation of TSH receptor (TSHR)-reactive B cells that produce TSHR-stimulating antibodies, manifesting as hyperthyroidism (“overactive thyroid”). In contrast, HT is characterized by apoptosis of thyroid cells, resulting in clinical hypothyroidism (“underactive thyroid”). Considerable progress has been made by many investigators to further our understanding of the mechanisms leading to thyroid autoimmunity. Although the exact etiology is not yet known, AITD are thought to arise from an interaction between genetic susceptibility factors, epigenetic effects, and various environmental triggers (e.g. iodine, infection), which leads to breakdown of immune tolerance. While the environmental exposures triggering AITD are still to be clarified, the genetic contributions to AITD are gradually being deciphered at the molecular level and are now more clearly understood. To date, there are at least seven genes that have been shown to contribute to the etiology of AITD (immune-regulatory genes (HLA-DR, CD40, CTLA-4, PTPN22, CD25, Tg, and TSHR) [for a review see Tomer Y. Annu Rev Pathol 2014; 9: 147-156]. There is now evidence that each gene alone exerts weak effects, but together these genes interact to produce stronger, synergistic effects, ultimately triggering disease. With recent advances in genomic sequencing and analyses tools, and the completion of the HapMap project and the 1000 genome project, these developments will help provide greater insight into the pathogenesis of thyroid autoimmunity.
AITD DEVELOP THROUGH A GENETIC-ENVIRONMENTAL
INTERACTIONS MEDIATED BY EPIGENETIC CHANGES
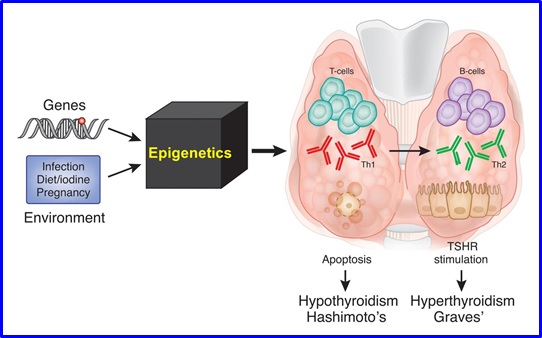
Environmental Triggers of Autoimmune Thyroid Diseases (AITD)
There have been many environmental exposures shown to be associated with the development of AITD. These include iodine (excess or deficiency), medications (e.g. interferon-alpha (IFN alpha) and Amiodarone), bacterial and viral infections (e.g. Yersinia Enterocolitica and hepatitis C virus), smoking, stress, and pollutants (e.g. polyaromatic hydrocarbons) (reviewed in Cocks Eschler et al. Clin Rev Allerg Immunol 2011; 41: 190-197). Potential mechanisms by which these agents induce AITD may involve interference with thyroid function, direct toxic effects on thyroid cells, and/or immune stimulation (Brent GA. Thyroid 2010; 20: 755-761). For example, iodine, while essential for normal thyroid function, could induce autoimmune thyroiditis by increasing immunogenicity of a highly iodinated thyroglobulin molecule, or by releasing free oxygen radicals as a result of direct tissue toxicity (see Papanastasiou et al. Thyroid 2007; 17: 729-739, and Lombardi et al. JCEM 2015; 100: E1-E10). Although initial data were conflicting, recent studies have substantiated hepatitis C virus (HCV) as an important infectious trigger of AITD. In addition, Interferon alpha, used to treat certain cancers, is recognized to be strongly associated with the development of thyroiditis (Mandac et al. Hepatology 2006; 43: 661-672). While the exact mechanisms by which these and other environmental factors interact with susceptibility genes to cause AITD emerging evidence from our lab and other labs strongly suggest that this interaction develops through epigenetic modifications of susceptibility genes by the environmental exposure.
Susceptibility Genes That Predispose To Autoimmune Thyroid Diseases
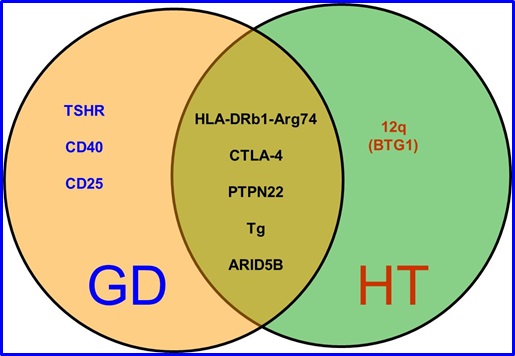
Epidemiological studies have long recognized the important role of genetics in the etiology of AITD. Family studies have shown that AITD cluster in families, sibling studies have shown a high sibling risk ratio for AITD, and twin studies have shown a significantly higher concordance rates for AITD in monozygotic twins compared to dizygotic twins (reviewed Tomer and Huber. J Autoimmun 2009; 32: 231-239). The first gene found to be associated with both GD and HT, was HLA-DR3 (Jacobson, Huber, Tomer. J autoimmune 2008; 30: 58-62). Since this discovery, significant progress has been made into the genetic contributions and the mechanisms underlying thyroid autoimmunity. To date several loci (genetic regions) and genes have been associated with AITD. In addition to HLA-DR3, these include 2 groups of non-HLA genes: (1) immune-regulatory genes (CD40, CTLA-4, PTPN22, FOXP3, and CD25); (2) thyroid-specific genes (Tg and TSHR).
SUSCEPTIBILITY GENES FOR GD AND HT
CURRENT PROJECTS IN THE TOMER LAB
Genetic Studies In Autoimmune Thyroid Diseases (AITD)
Our lab was the first to complete a whole genome linkage study in families in which AITD clustered. Our genome-wide approach mapped several new AITD loci and genes. Current projects focus on 2 AITD genes: CD40 and thyroglobulin:
- CD40: Our whole genome linkage study mapped a locus on chromosome 20q that was strongly linked with Graves’ disease (designated GD-2). Fine mapping and sequencing of this locus identified CD40, a member of the TNF Receptor family, to be the GD susceptibility gene in the 20q locus (Tomer et al. Am J Hum Genet 2003; 73: 736-747). Following our initial discovery that CD40 is a major Graves’ disease (GD) gene CD40 has now been confirmed as a key gene in several autoimmune diseases (e.g. rheumatoid arthritis, SLE, multiple sclerosis) and it is now clear that CD40 is a major autoimmunity gene. To identify the causative variant within CD40 that triggers GD we sequenced CD40 and identified the causative SNP as a 5’UTR SNP in the Kozak sequence of CD40. We are now studying the mechanisms by which the CD40 Kozak sequence SNP predisposes to GD. Our studies showed that the CD40 Kozak sequence SNP increases the translational efficiency of CD40 suggesting that the SNP predisposes to GD by increasing the expression of CD40 on antigen presenting cells and/or thyroid cells. Indeed, transgenic over-expression of CD40 in the thyroid accelerated GD in a mouse model of GD (experimental autoimmune GD [EAGD]) through an IL-6 dependent mechanism (Huber et al. J Immunol 2012; 189: 3043-3053). More recently we dissected the CD40 signaling pathway in thyroid cells that triggers GD and discovered that both the canonical and non-canonical NF-kB pathways are involved leading to cytokine production & inflammation (Lee et al. Endocrinology 2017; 158: 410-418). Currently our group continues to analyze the role of CD40 in triggering GD using novel mouse models.
- Thyroglobulin (Tg): Our GWLS mapped a locus on chromosome 8q that was linked with AITD. We later mapped the thyroglobulin (Tg) gene as the AITD susceptibility gene in the 8q locus. To identify the causative variant/s we sequenced the Tg gene, and identified specific amino acid substitutions in Tg that are strongly associated with AITD (Ban et al. PNAS 2003; 100: 15119-15124). Moreover, we showed an interaction between the key Tg amino acid substitution (W1999R) and an HLA-DR variant we identified (see below) giving a combined relative risk for GD of over 15. This statistical gene-gene interaction was followed by functional studies identifying the key Tg peptides (epitopes) that trigger AITD (see below). Currently our group is studying how to translate these findings into novel therapies for AITD.
MOST AITD SUSCEPTIBILITY GENES (CIRCLED IN RED) PARTICIPATE IN
ANTIGEN PRESENTATION
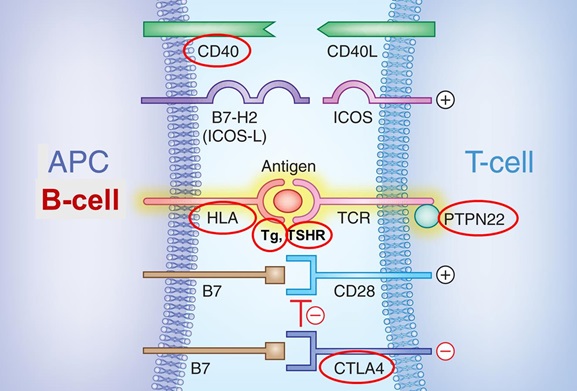
Dissecting the Role Of The HLA Locus In AITD
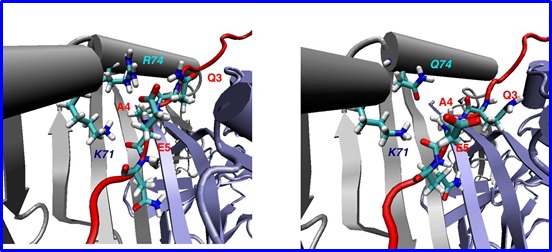
HLA-DR3 has been known to be the key HLA allele associated with GD and HT for nearly 40 years. However, HLA-DR3 has over 30 subtypes, and by sequencing the HLA-DRb1 gene we discovered the key HLA-DR pocket amino acid for the development of both GD & HT (arginine at position 74 of the HLA-DRb1 chain, designated DRb1-Arg74) (Menconi et al. PNAS 2008; 105: 14034-14039). This finding enabled us to identify the major thyroglobulin (Tg) epitope that triggers AITD. Using recombinant HLA-DR3 containing the DR variant specific for AITD (HLA-DRb1-Arg74) we screened the Tg protein for peptides that bind to the DRb1-Arg74 pocket identifying 5 strong binders (Jacobson et al. JBC 2009; 284: 34231-34243). Of these, one peptide, Tg.2098 also triggered T-cell responses in humanized mice expressing human DRb1-Arg74 and in patients with AITD. These findings have significant translational implications as they open the way to identifying compounds that can block the binding of Tg.2098 to HLA-DRβ1-Arg74 and its presentation to T-cells (see below). This is now a major research focus of our lab.
3 DIMENSIONAL STRUCTURE OF HLA-DRB1-Arg74
THYROGLOBULIN PEPTIDE CAN BIND TO THE ARGININE 74 (SUSCEPTIBLE
VARIANT OF HAL-DR) AND NOT TO THE GLUTAMINE 74 (PROTECTIVE
VARIANT OF HLA-DR)
3 DIMENSIONAL STRUCTURE OF HLA-DRB1-Arg74
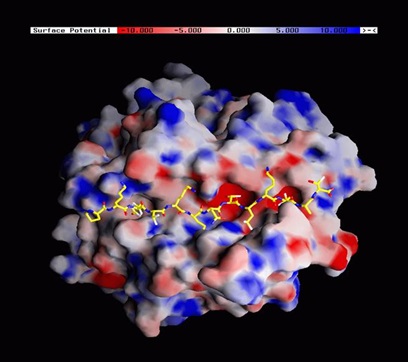
THYROGLOBULIN PEPTIDE CAN BIND TO THE ARGININE 74 (SUSCEPTIBLE
VARIANT OF HAL-DR) AND NOT TO THE GLUTAMINE 74 (PROTECTIVE VARIANT OF HLA-DR)
Identifying the joint susceptibility genes for AITD and type 1 diabetes (T1D)
Our group was the first to performed a whole genome linkage study in families in which both AITD and type 1 diabetes (T1D) cluster. Our linkage study identified new genes and loci that are linked with AITD and T1D. We showed that CTLA-4 and FOXP3 are key susceptibility genes for AITD+T1D (APS3v) (Villano et al. JCEM 2009; 94: 1458-1466). Recently we used a genome-wide approach to identify new genes in patients that have both autoimmune thyroiditis and type 1 diabetes (a variant of the autoimmune polyglandular syndrome type 3, designated APS3v). Our studies identified new susceptibility genes for AITD+T1D (APS3v), specifically GPR103 (Tomer et al. J Autoimmun 2015; 60: 32-39). In addition, we identified a unique HLA-DR pocket amino acid signature that confers a high risk for the development of autoimmune thyroiditis and type 1 diabetes in the same patient (APS3v) (Menconi et al. PNAS 2010; 107: 16899-16903). Structural analysis showed that this amino acid signature creates a unique HLA-DR pocket structure enabling the presentation of pathogenic peptides. Moreover, we recently identified the 4 key peptide epitopes that trigger AITD+T1D in the same individual (APS3v) (Li et al. J Autoimmun 2017; 76: 1-9). Current projects in the lab focus on identifying new non HLA variants that predispose to APS3v and translating these findings into novel therapies in preclinical studies using mouse models.
Epigenetic-Mechanistic Studies in AITD and Type 1 Diabetes (T1D)
Recently there was a paradigm shift in our understanding of the mechanisms by which genetic variants trigger complex diseases. Four seminal papers published in 2013 [Kilpinen et al Science 2013; 342: 744-747, McVicker et al. Science 2013; 342: 747-749, Kasowski et al Science 2013; 342: 750-752; Heinz et al Nature 2013; 503: 487-492] showed that certain non-coding SNP alleles can determine phenotype (including disease) by facilitating transcription factor binding to regulatory gene regions leading to histone modifications that alter gene transcription. Our work has shown that this type of genetic-epigenetic interactions can trigger autoimmune thyroid diseases by affecting the transcription of the Tg and TSHR genes.
- Thyroglobulin (Tg) gene: In 2011 we identified a SNP in the thyroglobulin gene (Tg) promoter that interacts epigenetically through histone modifications with IRF-1, a transcription factor induced by interferon alpha during infections. The interaction of IRF-1 with the G (risk) allele of the Tg SNP altered Tg gene expression which in the setting of inflammation can trigger thyroid autoimmunity (Stefan et al. JBC 2011; 286: 31168-31179). Current projects in the lab are aimed at identifying the exact mechanisms by which the upregulation of Tg expression induced by this genetic-epigenetic interaction triggers AITD.
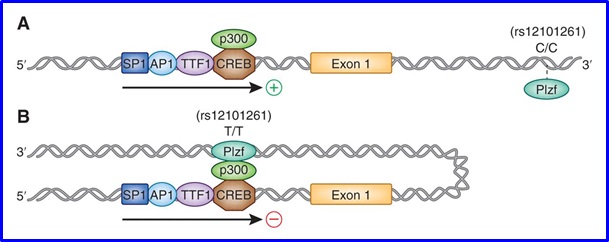
- Analyzing the TSHR gene: We discovered a new genetic-epigenetic interaction between a SNP in intron 1 of the TSHR gene and the transcription factor - PLZF. This genetic-epigenetic interaction led to reduced thymic expression of TSHR. Reduced thymic expression of TSHR can lead to escape from central tolerance and trigger Graves’ disease (Stefan et al. PNAS 2014; 111: 12562-12567). Current projects in the lab are aimed at pipointing the mechanisms by which this genetic-epigenetic interaction leads to an autoimmune response targeting the TSHR.
THE TRANSCRIPTION FACTOR PLZF CAN ONLY BIND TO THE BINDING ELEMENT IN
INTRON 1 OF THE TSHAR WHEN THE TT (SUSCEPTIBLE) GENOTYPE IS PRESENT AND NOT THE C/C (PROTECTIVE) GENOTYPE. BINDING OF PLZF TO THE BINDING ELEMENT IN INTRON 1 INHIBITS TSHR TRNASCRIPTION
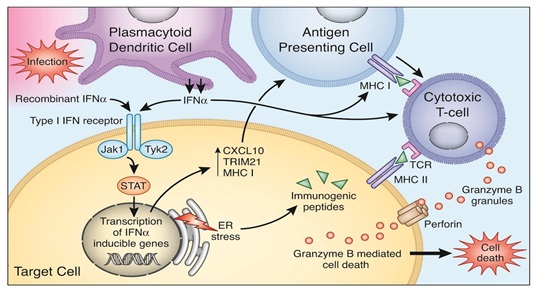
- Studies in type 1 diabetes (T1D): We have shown that genetic–epigenetic interactions are also key to the development of type 1 diabetes (T1D). Through studies of twins with T1D we demonstrated that changes in DNA methylation at the HLA, Insulin, IL-2RB and CD226 genes may play an important role in the etiology of type 1 diabetes (Stefan et al. J Autoimmun 2014; 50: 33-37). Current projects in the lab are aimed at dissecting the effects of DNA methylation changes at these genes on their function and how it can trigger T1D. Another focus of our lab is on the role of interferon alpha in triggering AITD and type 1 diabetes. Interferon alpha is the key cytokine secreted during viral infections and has been implicated in triggering AITD and T1D (See our manuscript Akeno et al. J Immunol 2011; 186: 4693-4706). Our Studies have shown that interferon alpha induces endoplasmic reticulum (ER) stress in human beta cells, and interfers with insulin production and secretion (Lombardi and Tomer. J Autoimmun 2017; 80: 48-55). Current projects in the lab focus on dissecting the mechanisms by which interferon alpha induces changes in thyroid cells and pancreatic islet beta cells that lead to the development of AITD and T1D, respectively.
SCHEMATIC REPRESENTATION OF MECHANISMS BY WHICH INTERFERON
ALPHA CAN TRIGGER AUTOIMMUNE THYROIDITIS
Translational studies in AITD and T1D
Our group has been translating our discoveries into novel therapeutic approaches in
Graves’ disease, Hashimoto’s thyroiditis and type 1 diabetes. Our novel therapeutic approach is based on blocking antigen presentation. This therapeutic approach is based on our discovery of the unique HLA-DR pocket (DRb1-Arg74) that predisposes to Graves’ disease and Hashimoto’s thyroiditis (Menconi et al. PNAS 2008; 105: 14034-14039) and the identification of the specific thyroglobulin peptides that bind to the DRb1-Arg74 pocket to trigger AITD (Jacobson et al. JBC 2009; 284: 34231-34243). We have now used this knowledge to develop novel therapies for AITD by blocking the DRb1-Arg74 pocket. Indeed, recently our studies discovered that a compound called Cepharanthine can block the HLA-DRb1-Arg74 pocket (Li et al. JBC 2016; 291: 4079-4090). Current studies in the lab are aimed at studying whether Cepharanthine can block the autoimmune response to thyroglobulin in AITD.




